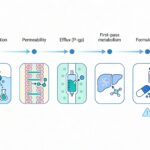
Bioavailability (F) is formally defined as the fraction of an administered dose of an active pharmaceutical ingredient (API) that reaches the systemic circulation in an unchanged form, integrating both absorption efficiency and the impact of first-pass metabolism. For orally administered compounds, bioavailability is governed by the interplay of physicochemical properties, gastrointestinal physiology, transport processes, intestinal permeability, efflux transport mechanisms, and presystemic metabolism. Suboptimal bioavailability remains one of the dominant causes of attrition in drug development and a persistent challenge in clinical translation.
Mechanistic determinants of poor bioavailability
1. Dissolution-limited absorption
For solid oral dosage forms, dissolution is a prerequisite for absorption. According to the Noyes–Whitney equation, dissolution rate is directly proportional to surface area and concentration gradient, and inversely proportional to diffusion layer thickness. Many contemporary drug candidates exhibit high lipophilicity and crystalline stability, resulting in poor aqueous solubility, dissolution-limited absorption, and slow dissolution kinetics.
This issue is particularly prevalent among Biopharmaceutics Classification System (BCS) Class II and IV compounds, where absorption is solubility-limited rather than permeability-limited. In such cases, gastrointestinal transit time becomes a critical bottleneck, as incomplete dissolution leads directly to reduced systemic exposure and low oral bioavailability.
This phenomenon makes dissolution‑limited absorption one of the central challenges in bioavailability optimization.
2. Limited intestinal permeability
Even when dissolution is adequate, transcellular or paracellular transport across the intestinal epithelium may be inefficient. Intestinal permeability is influenced by:
- Molecular size and polarity
- Hydrogen bonding capacity
- Lipophilicity–polarity balance (logP/logD)
Compounds with unfavorable permeability profiles fail to achieve sufficient flux across enterocytes, resulting in incomplete absorption despite adequate luminal concentration. This limitation is particularly relevant for BCS Class III and IV molecules, where permeability rather than solubility becomes the dominant barrier.
3. Chemical and enzymatic instability
Certain APIs are labile under gastric or intestinal conditions, undergoing degradation due to:
- Acid-catalyzed hydrolysis
- Enzymatic cleavage by luminal or brush-border enzymes
Such degradation reduces the fraction of intact compound available for absorption, effectively lowering apparent bioavailability independent of permeability considerations. In these cases, enteric coatings and protective formulation strategies are often required to preserve molecular integrity.
4. Presystemic (first-pass) metabolism
Following intestinal absorption, portal circulation delivers drug molecules directly to the liver, where cytochrome P450 enzymes, conjugation pathways, and hepatic uptake transporters can extensively metabolize the compound prior to systemic distribution.
Additionally, intestinal metabolism, particularly mediated by CYP3A4 and UGT enzymes, may significantly contribute to presystemic clearance. For drugs with high intrinsic clearance, first-pass metabolism can reduce oral bioavailability to single-digit percentages despite efficient absorption.
5. Efflux transport mechanisms
Efflux transporters such as P-glycoprotein (ABCB1) and BCRP (ABCG2) are highly expressed in enterocytes and actively limit intracellular drug accumulation by exporting substrates back into the intestinal lumen.
This phenomenon creates a functional barrier to absorption, particularly for lipophilic and structurally complex molecules, and often operates synergistically with intestinal metabolism to further reduce systemic exposure. Modulation of efflux transporters is therefore an important consideration in bioavailability optimization.
Formulation science as a bioavailability-enabling strategy
Modern pharmaceutical development increasingly treats bioavailability as a formulation-dependent variable rather than an immutable property of the API, leveraging advances in solubility enhancement, permeability modulation, and mitigation of first-pass metabolism.
Solubility enhancement strategies
Advanced formulations improve dissolution kinetics and apparent solubility through:
- Particle size reduction (micronization, nanocrystals)
- Amorphous solid dispersions
- Lipid-based delivery systems
These approaches increase thermodynamic activity and maintain supersaturation in the gastrointestinal environment, thereby enhancing absorptive flux and improving overall oral bioavailability.
Protection against degradation
Enteric coatings, encapsulation technologies, and carrier systems are employed to:
- Shield labile APIs from acidic or enzymatic degradation
- Enable site-specific release in regions of optimal absorptive capacity
This preserves molecular integrity until absorption can occur and reduces preabsorptive loss due to chemical or enzymatic instability.
Permeability and transport modulation
Formulation strategies may exploit physiological lipid absorption pathways or transiently modulate membrane interactions to facilitate transcellular transport. In some systems, excipients are selected to influence transporter activity, reduce efflux, and enhance intestinal permeability or alter microenvironmental conditions at the epithelial surface.
Mitigation of first-pass metabolism
Certain delivery systems alter the rate and route of absorption, promoting lymphatic uptake or prolonged input into systemic circulation, thereby reducing hepatic extraction. Controlled release profiles may also lower peak portal concentrations, decreasing metabolic saturation and clearance.
Clinical and translational implications
From a clinical pharmacology perspective, bioavailability directly impacts:
- Dose requirements
- Interindividual variability
- Exposure–response relationships
- Therapeutic index and safety margins
Formulation-driven bioavailability enhancement can transform a marginal compound into a clinically viable therapy, underscoring the critical role of pharmaceutical technology in drug efficacy, safety, and reproducibility.
Summary
Poor bioavailability arises from a convergence of unfavourable physicochemical properties, biological barriers, and metabolic processes. Contemporary formulation science addresses these challenges by strategically modifying drug presentation to align with gastrointestinal physiology and systemic disposition mechanisms. As a result, bioavailability is no longer merely a limitation—but a design parameter in modern drug development, shaped by solubility enhancement, permeability optimization, efflux modulation, and mitigation of first-pass metabolism.
Further reading: Enhancing Oral Drug Absorption: Overcoming Physiological and Pharmaceutical Barriers for Improved Bioavailability
If you’re exploring this topic for the first time: